
Researchers using the BioCARS sector 14 beamline at the APS have determined how changes in a pair of proteins lead to the family of neurological disorders that includes Tay-Sachs disease. They solved the three-dimensional structure of one protein essential to Tay-Sachs and modeled the structure of a second key protein based on the first. The results help explain why different mutations lead to different forms of the same class of neurological disorder, and may help in developing treatments against them.
Tay-Sachs is caused by an inherited deficiency of the enzyme Hex A, which removes a sugar residue from fatty molecules found in brain cells. Without it, fat builds up in the cells and kills them in a still mysterious way. Three genes must interact to remove the sugar properly: HEXA and HEXB, which produce the a- and b-subunits of Hex A, respectively; and GM2A, whose protein product, Gm2A, activates Hex A. Mutations that significantly reduce the activity of these genes can cause one of several related disorders, including Tay-Sachs and Sandhoff disease. One in 35 Ashkenazi Jews carries the mutation that causes Tay-Sachs, and 1 in 300 individuals in general carries it. Understanding the structures of the proteins involved might enable researchers to design drugs to activate or replace them. However, researchers had, to date, only determined the structure of the Gm2A protein.
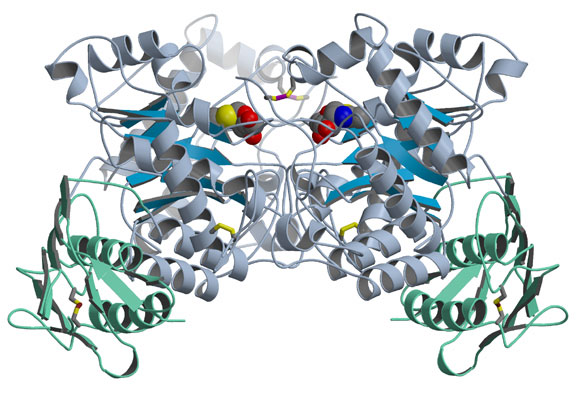
The group solved two structural problems at once - Hex A and the closely related Hex B - as reported in the April 11, 2003, issue of the Journal of Molecular Biology. They first crystallized Hex B, made of two b-subunits, and determined its structure by analyzing its x-ray diffraction pattern. In Hex B, which is easier to crystallize than Hex A, two kidney-shaped b-subunits come together so their active sites, or catalytic regions, face each other but are offset (Fig. 1). Previous studies had indicated that each type of Hex subunit has an active site capable of performing a slightly different type of sugar removal, but that two subunits must come together for either one to be active. To determine how the b-subunit active site binds its substrate, the researchers soaked a Hex B inhibitor that mimics the substrate into the crystallized enzyme. They found that the active sites work cooperatively because several of the amino acids that make up each subunit's active site come from the other subunit. People who have mutations in these amino acids experience a mild, chronic form of Sandhoff disease, or a variant or acute form of Tay-Sachs, depending on which amino acid is mutated. Soaking the crystal with a different inhibitor also confirmed that Hex B uses a similar catalytic mechanism to related enzymes.
Next they modeled the a-subunit based on the b-subunit, which has 60% of the same amino acids. In support of the modeling approach, much of the amino acid similarity occurs in the regions implicated in subunit joining or catalysis, suggesting that the b-subunit binds the a in the same way it binds itself; Hex A would probably need a similar overall shape to that of Hex B to support the same type of subunit binding. The model indicates that three amino acids in the a-subunit are responsible for Hex A's ability to bind substrates with a negative charge at one end. Biochemical studies of the enzyme's reaction rate when these amino acids are mutated support the conclusion. The predicted subunit interface also forms a large groove suitable for binding the cup-shaped activator protein Gm2A. Shape and electrostatic complementarities suggest that the activator positions itself over the a-subunit active site.
See: B.L. Mark 1, D.J. Mahuran 2,3, M.M. Cherney 1, D. Zhao 4, S. Knapp 4, and M.N.G. James 1, "Crystal Structure of Human b-Hexosaminidase B: Understanding the Molecular Basis of Sandhoff and Tay-Sachs Disease," J. Mol. Biol. 327, 1093-1109 (2003).
Author affiliations: 1University of Alberta, 2The Hospital for Sick Children, 3University of Toronto, 4Rutgers University
This work supported by Canadian Institutes of Health Research grants (to D.M. and M.N.G.J.). B.L.M. is supported by scholarships from the C.I.H.R., the Alberta Heritage Foundation for Medical Research, and the University of Alberta. M.N.G.J. holds a Canada Research Chair in Protein Structure and Function at the University of Alberta. Use of the BioCARS beamline was supported by the National Institutes of Health, National Center for Research Resources, under grant number RR07707. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-ENG-38.