
|
In this example, a single crystallographic phase will be used to model both the nuclear and magnetic scattering. The phase will be set in a supercell (Fmmm) of the actual nuclear cell (Pmmm) so some constraints will be needed. The nuclear scattering will be modeled first and then magnetic scattering will also be included in the computation. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Input model for Nuclear Scattering Phase | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
The first step in the refinement is to start a new refinement by creating a new experiment file. Start EXPGUI and enter a new file name then click on the "Create" button to then create a new, empty, experiment file (screen image) and enter a title. The first step in creating an GSAS refinement is to create a phase. This can be done from this information:
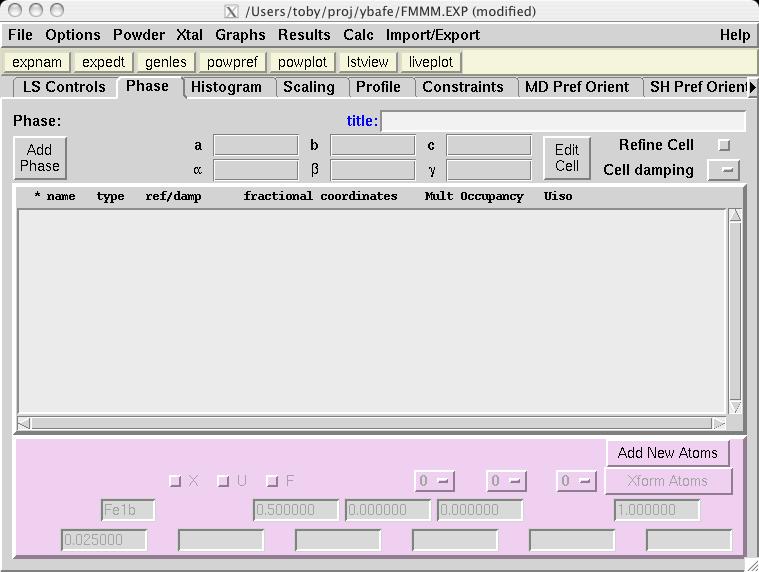
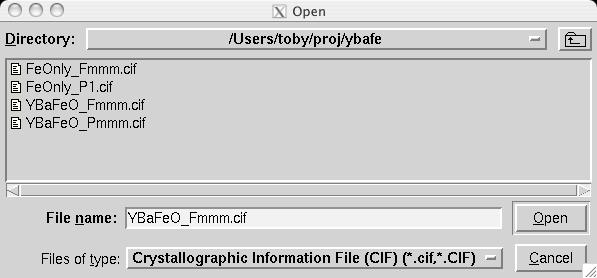
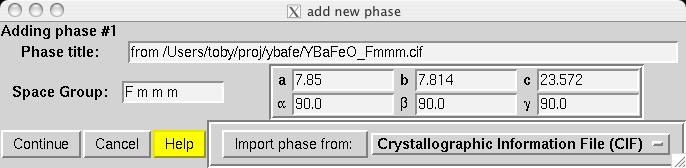
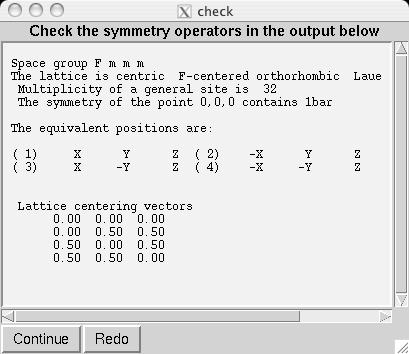
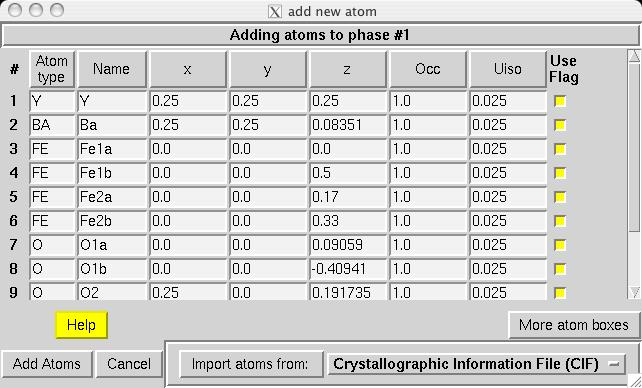
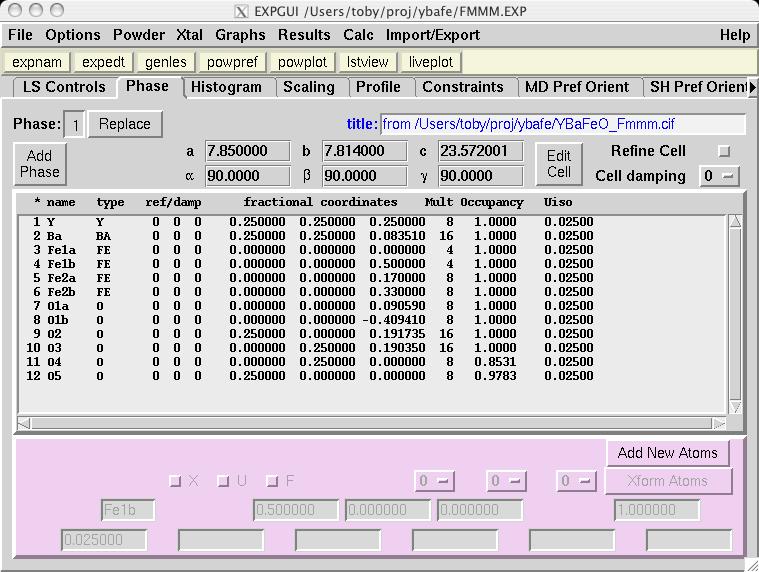
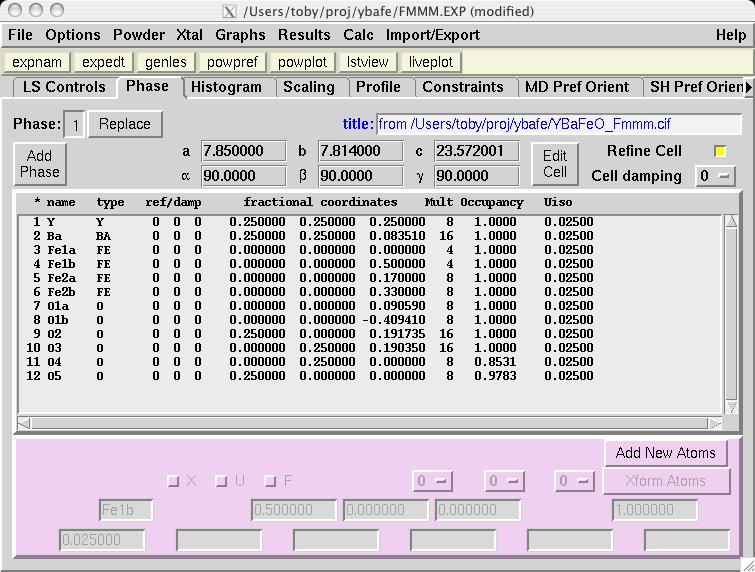
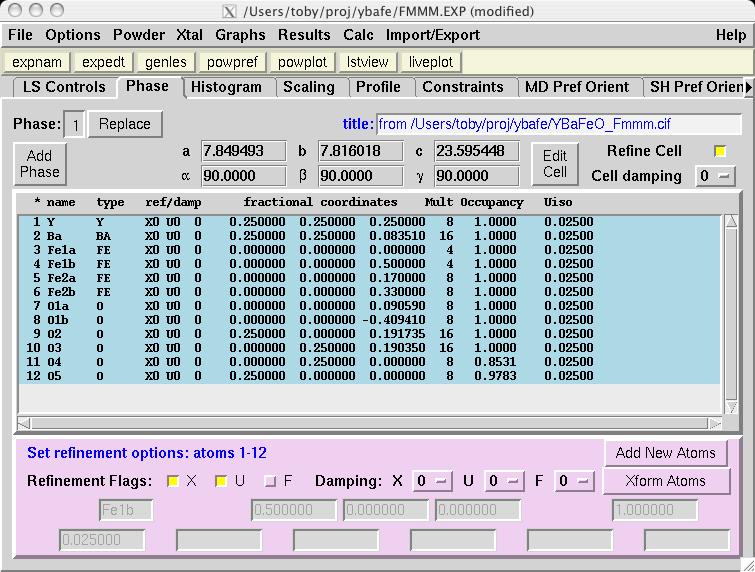
Note that in comparison to the Pmmm structure, the Fe1, Fe2 and O1 atoms are included twice and thus should be constrained to retain their symmetry. Also, several parameters that were previously fixed due to symmetry, x(O2), y(O3), y(O4) and x(O5), are now on lower symmetry positions where the value 0.25 is not fixed by symmetry. This must be included in the model manually. Considerable typing can be avoided by importing the phase from a provided CIF, YBaFeO_Fmmm.cif in file YBAFEO.zip rather than by typing in the information shown in the screen images in the following sections. Go to the Phase panel (screen image) and press the "Add Phase" button. To input the information from the CIF, click on the "PowderCell .CEL file" menu button to bring up a menu of choices (screen image) and select "Crystallographic Information File (CIF)" which brings up a window where the file YBaFeO_Fmmm.cif can be selected. (screen image); click on "Open" after the file is selected. Click on "Continue" on the add new phase (screen image) and on the Check symmetry windows (screen image) and then "Add Atoms" on the Adding atoms... window (screen image) and the phase has been added, just like that. Finally, make sure the "Refine Cell" option is not selected, (screen image) so that the unit cell parameters will be refined until later, after the background and scale factor are first brought into the right range. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Input Diffraction Data | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
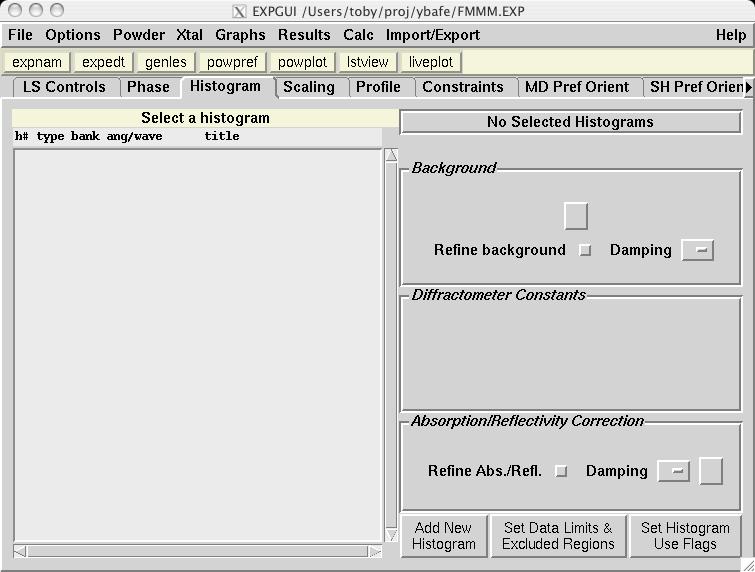
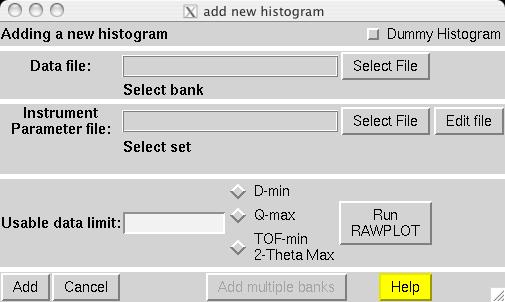
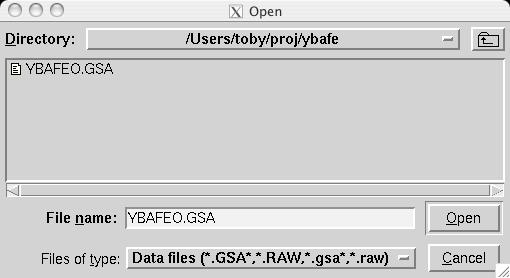
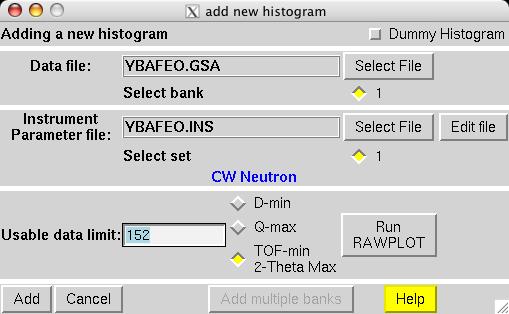
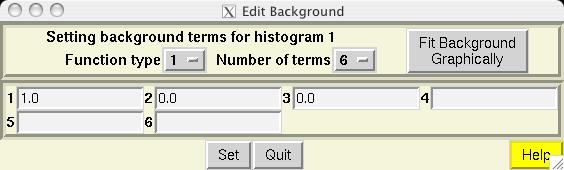
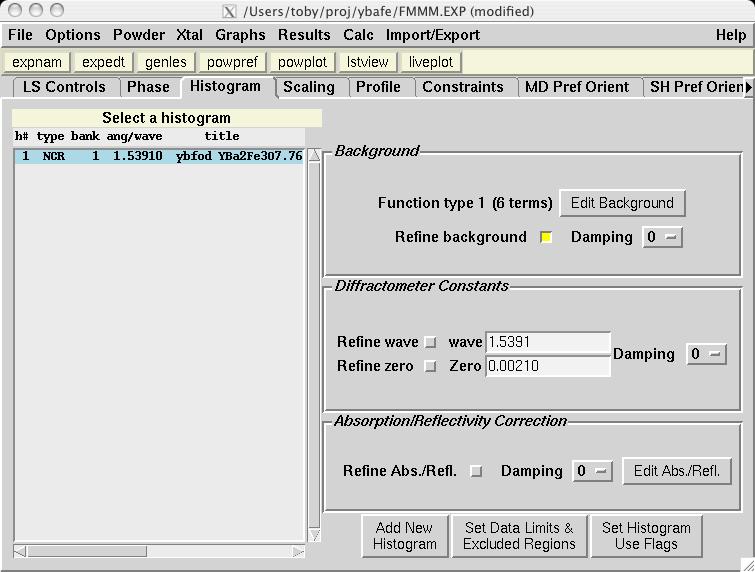
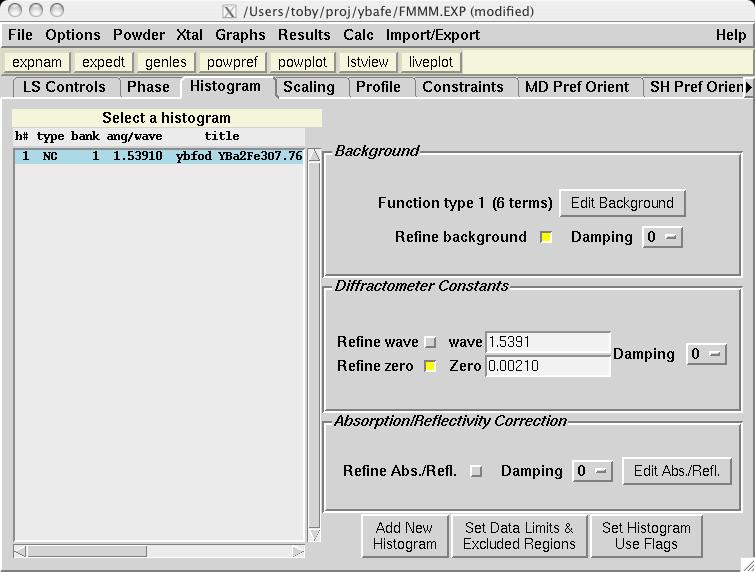
The second step in almost every GSAS refinement is to input diffraction data (or select data parameters in a simulation). This is done from the Histogram panel (screen image). Click on the "Add New Histogram" button which brings up the add new histogram window (screen image). Press the Select file to select the appropriate GSAS data file in the Open window (screen image); press the open button and the file names for the both the data file and the instrument parameter files are loaded (you might see messages about file conversions if the download process has stripped non-printing characters needed from the files.). Finally, change the data range to utilize a maximum two-theta of 152 degrees on the add new histogram window, which eliminates the last very wide peak from use in the refinement (the fit would also progress well were this peak included) (screen image). Press "Add" on this window and the diffraction data are now included in the refinement Finally, change the background function to type 1 with 6 terms by pressing the "Edit Background" button (this is the authors' preference -- but other settings could be selected.) (screen image). Finally, confirm that the "Refine background" option is selected so that the background will be fit (screen image). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Start Initial Fit of Experimental Parameters | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
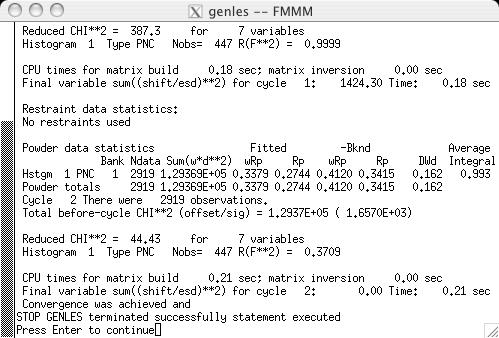
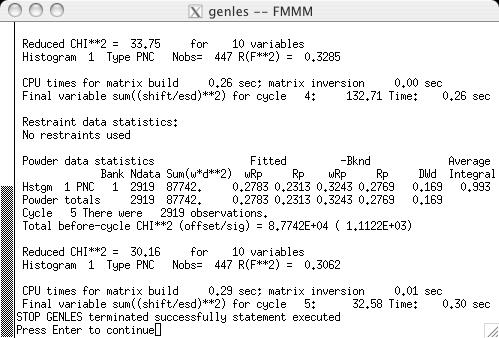
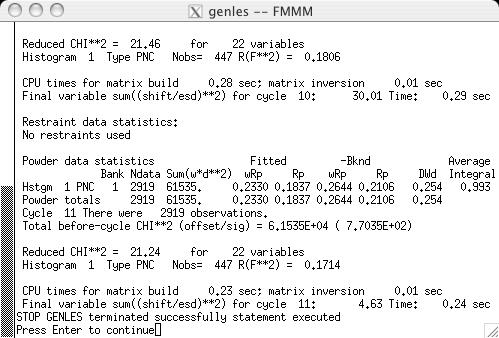
The fit can now be started, by pressing the "powpref" (screen image) and "genles" buttons (screen image). Note that pressing each button starts a program in another window and then (unless the default settings have been changed) after each run has been completed, the extra window is closed and the "Load New" button is pressed on the Reload screen (screen image). Note that after the two cycles of refinement, the chi2 value drops to approximately 44. At this point the unit cell is refined, by selecting the the "Refine Cell" option on the Phase panel (screen image). Press the "genles" button and note that the fit improves to a chi2 value of approximately 30 (screen image). The two-theta zero is now refined using the "Refine zero" control on the Histogram panel (screen image). Press the "genles" button and note that the fit improves to a chi2 value of approximately 28 (screen image). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Constrain Structural Parameters | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
At this point an initial fit of the structure is performed (although some researchers might delay this until the magnetic modeling is included). While under normal conditions, it would be wise to add refinement of variables over the course of several refinement runs, in this case the model is quite stable with respect to refinement and this can be done in a single run. Before starting the refinement, however, Uiso values for like element types are grouped together to reduce the number of parameters. This is done with the Constraints panel (screen image).
As mentioned previously, the 0.25 values for O atom coordinates are not special values in Fmmm and must be constrained. This must be done with EXPEDT using the following commands:
where command 1) [Y] indicates the correct file has been selected to be edited; command 2) [L] enters the least-squares menu and then [A] moves from the least-squares to the atoms edit submenu; command 3) [F] moves to the fix parameters submenu; command 4-7) [i N x/y] fixes the x or y parameter of atom N, so that this value does not change when the atom position is varied. command 8) [L] lists the held parameters in the current phase (#2) -- this command is optional; command 9) [x x x x] returns to the main menu; command 10) [x] exits EXPEDT. The input to and output from EXPEDT are shown below. User-typed input is emphasized by display in this font: Input
|------------------------------------------|
| Program EXPEDT Version MacOSX |
| A menu driven routine to edit .EXP files |
| Distributed on Thu Sep 30 14:58:36 2004 |
|------------------------------------------|
|---------------------------------------------------------------|
| Allen C. Larson and Robert B. Von Dreele |
| Manuel Lujan, Jr. Neutron Scattering Center, MS-H805 |
| Los Alamos National Laboratory, Los Alamos, NM 87545 |
| |
| Copyright, 2000, The Regents of the University of California. |
|---------------------------------------------------------------|
The last history record is :
HSTRY 13 EXPGUI 1.74 1.42 (4 changes) -- 07/30/05 20:46:38
Is this the file you wish to use? (,D,K,Q,R,Y) >Y
Experiment title:
Model nuclear & magnetic scattering in Fmmm
The last history record is :
HSTRY 13 EXPGUI 1.74 1.42 (4 changes) -- 07/30/05 20:46:38
EXPEDT data setup option (,D,F,K,L,P,R,S,X) >L A
Phase No. 1; Phase has 12 atoms; Title: from /Users/toby/proj/ybafe/YBaFeO_
Give atom editing command
(,$,C,D,E,F,I,K,L,M,S,T,U,V,X,+,-,*,/) >F
There currently no parameters being held for this phase
Enter option (,D,I,L,X) >i 9 x
Enter option (,D,I,L,X) >i 10 y
Enter option (,D,I,L,X) >i 11 y
Enter option (,D,I,L,X) >i 12 x
Enter option (,D,I,L,X) >l
Parameters being held constant are
1(1 9X ) 2(1 10Y ) 3(1 11Y ) 4(1 12X )
Enter option (,D,I,L,X) >x x x x
Phase No. 1; Phase has 12 atoms; Title: from /Users/toby/proj/ybafe/YBaFeO_
EXPEDT data setup option (,D,F,K,L,P,R,S,X) >x
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Start Initial Fit of Structural Parameters | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
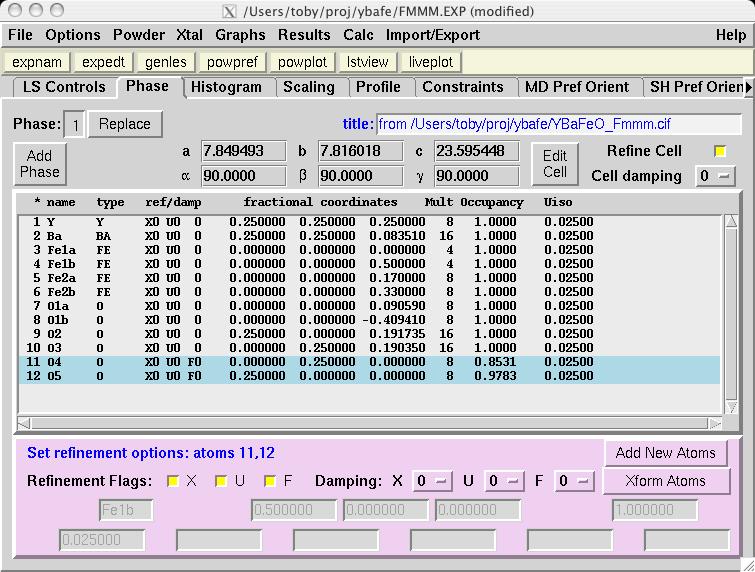
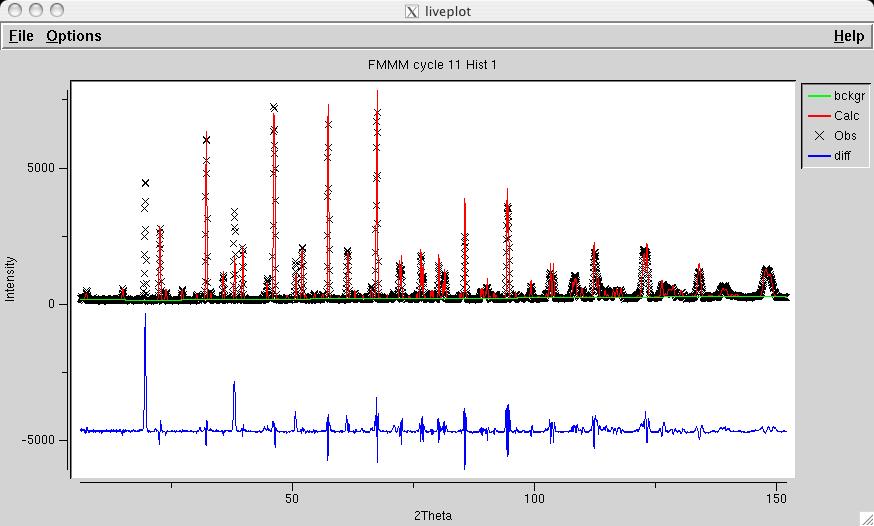
To refine the nuclear model, select all atoms on the phase panel (the right mouse button will do this) and click on the X and U "Refinement Flag" buttons (screen image) so that all atom positions can be optimized in addition to 4 Uiso values (1 for the Fe atoms, 1 for the O atoms, 1 for the Y atom and 1 for the Ba atom). Then set the F (fractional occupancy) flag for O4 and O5 only (screen image). Press the "genles" button and note that the fit improves to a chi2 value of approximately 21 (screen image). A view of the fit in LIVEPLOT shows the profile terms need some refinement (which will be done later) and there is significant missing intensity for several low angle reflections (screen image) -- not surprising, since magnetic scattering is not included. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Include Magnetic Scattering | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
At this time, EXPGUI does not implement many of the controls needed for magnetic structure fitting. Nonetheless, as will be shown here. it is still possible to use EXPGUI for many steps, and then use EXPEDT for steps that cannot be done at present in EXPGUI. To add magnetic scattering, EXPEDT is used to change a flag so the phase is recognized as both a magnetic as well as nuclear scatterer. The EXPEDT program is invoked from EXPGUI by pressing the EXPEDT button in the buttom bar or from the Powder and Xtal menus. To designate the phase as both a nuclear and magnetic scatter, the following commands are used in EXPEDT:
EXPEDT prompts the user to supply a one-letter command, where inputting a blank line provides further information on what the options are (as demonstrated below). Note that commands can be combined onto the same line (usually) and can in many cases be spread over more than one line. The input to and output from EXPEDT are shown below. User-typed input is emphasized by display in this font: Input
|------------------------------------------|
| Program EXPEDT Version MacOSX |
| A menu driven routine to edit .EXP files |
| Distributed on Thu Sep 30 14:58:36 2004 |
|------------------------------------------|
|---------------------------------------------------------------|
| Allen C. Larson and Robert B. Von Dreele |
| Manuel Lujan, Jr. Neutron Scattering Center, MS-H805 |
| Los Alamos National Laboratory, Los Alamos, NM 87545 |
| |
| Copyright, 2000, The Regents of the University of California. |
|---------------------------------------------------------------|
The last history record is :
HSTRY 16 GENLES MacOSX Jul 30 20:53:43 2005 Sdsq= 0.615E+05 S/E= 4.63
Is this the file you wish to use? (,D,K,Q,R,Y) >Y
Experiment title:
Model nuclear & magnetic scattering in Fmmm
The last history record is :
HSTRY 16 GENLES MacOSX Jul 30 20:53:43 2005 Sdsq= 0.615E+05 S/E= 4.63
EXPEDT data setup option (,D,F,K,L,P,R,S,X) >P
Select editing option for Powder data preparation (,H,P,T,X) >P
There is phase information present
Name of phase no. 1
from /Users/toby/proj/ybafe/YBaFeO_Fmmm.cif
The phase is non-magnetic
Enter phase edit command(,$,D,E,F,M,I,L,R,S,X) >M
Give phase number for phase type flag toggle >1
The phase is non-magnetic
Enter phase type (,A,B,C,D,L,X,Z) >
Selection of phase type:
A - Nuclear structure only
B - Nuclear and magnetic structure
C - Magnetic structure only
D - Macromolecular structure
Z - Pawley extraction for all phases
Enter phase type (,A,B,C,D,L,X,Z) >B
The phase is magnetic.
Both nuclear and magnetic peaks will be included when generating reflections fo
r powder patterns
Enter phase type (,A,B,C,L,X,Z) >x x x x
EXPEDT data setup option (,D,F,K,L,P,R,S,X) >
One more X command could now be entered to exit EXPEDT, but since the next step will continue in EXPEDT, this is not necessary. Note that the magnetic symmetry menu (as shown below) must be entered, even if no changes will be made, for GENLES to function once a magnetic phase has been added. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Setup Magnetic Symmetry and Moments | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
In the previous step, the phase was flagged as magnetic, but before a magnetic scattering computation can be performed, the magnetic symmetry needs to be set. In this case, two operator colors must be changed to change the space group from Fmmm to Fmm'm'. Once the magnetic symmetry is established, the magnetic moments for each magnetic atom are supplied. In EXPEDT, use the following commands:
Note that if EXPGUI were to be started at this point, the first command would be "Y" to indicate that the correct file has been selected to be edited. Command 1) [L] enters the least-squares menu and then [A] moves from the least-squares to the atoms edit submenu; command 2) [M] moves to the magnetism editing submenu; command 3) [S] enters the spin-flip sub menu; command 4) [C 2] toggles the color of the second symmetry operation (m perpendicular to y); command 5) [C 2] toggles the color of the third symmetry operation (m perpendicular to z); command 6) [x] returns to magnetism menu command 7-10) [m N M] sets the magnetic moment for atom N to M Bohr magnetrons. Note that in this case, these atoms are constrained by symmetry to have only one magnetic moment, in the x direction. If lower symmetry were present, components in other directions would be required. Command 11) [x] returns to the atoms edit menu; command 12) [L] lists the atoms in the current phase -- this command is optional; command 13) [x x x x] returns to the main menu. The input to and output from EXPEDT are shown below. User-typed input is emphasized by display in this font: Input
EXPEDT data setup option (,D,F,K,L,P,R,S,X) >L A
Phase No. 1; Phase has 12 atoms; Title: from /Users/toby/proj/ybafe/YBaFeO_
Give atom editing command
(,$,C,D,E,F,I,K,L,M,P,S,T,U,V,X,+,-,*,/) >M
Enter magnetism editing option (,A,C,L,M,S,X) >S
These changes may alter the allowed moment orientations and constraints.
All of the following data refer to the current space group symbol
Spin flip editing commands are
C n - Change spin flip "n"
L - List the current spin flip info
X - Exit to the atom magnetic data editing menu
Enter spin flip editing command (,C,L,X) >c 2
The unique symmetry operations and the associated spin colors are
Mx My Mz Acen Bcen Ccen
Black Red Black Black Black Black
Enter spin flip editing command (,C,L,X) >c 3
The unique symmetry operations and the associated spin colors are
Mx My Mz Acen Bcen Ccen
Black Red Red Black Black Black
Enter spin flip editing command (,C,L,X) >x
Enter magnetism editing option (,A,C,L,M,S,X) >m 3 3.5
Fe1a 0.000000 0.000000 0.000000
No magnetic moment is currently defined
Constraints on the moment are 1 1.00 0 0.00 0 0.00
New moment = 3.50000 0.00000 0.00000 3.500 90.000 0.000
Enter magnetism editing option (,A,C,L,M,S,X) >m 4 -3.5
Fe1b 0.000000 0.000000 0.500000
No magnetic moment is currently defined
Constraints on the moment are 1 1.00 0 0.00 0 0.00
New moment =-3.50000 0.00000 0.00000 3.500 90.000 180.000
Enter magnetism editing option (,A,C,L,M,S,X) >m 5 -3.5
Fe2a 0.000000 0.000000 0.169428
No magnetic moment is currently defined
Constraints on the moment are 1 1.00 0 0.00 0 0.00
New moment =-3.50000 0.00000 0.00000 3.500 90.000 180.000
Enter magnetism editing option (,A,C,L,M,S,X) >m 6 3.5
Fe2b 0.000000 0.000000 0.330573
No magnetic moment is currently defined
Constraints on the moment are 1 1.00 0 0.00 0 0.00
New moment = 3.50000 0.00000 0.00000 3.500 90.000 0.000
Enter magnetism editing option (,A,C,L,M,S,X) >x
Phase No. 1; Phase has 12 atoms; Title: from /Users/toby/proj/ybafe/YBaFeO_
Give atom editing command
(,$,C,D,E,F,I,K,L,M,P,S,T,U,V,X,+,-,*,/) >L
SER TYPE X Y Z FRAC NAME UISO CODE STSYM MULT FXU
MX MY MZ MCODE
1 Y 0.25000 0.25000 0.25000 1.00000 Y 0.00874 I XU 222 8 000
2 BA 0.25000 0.25000 0.08598 1.00000 Ba 0.01200 I XU 2(001) 16 000
3 FE+3 0.00000 0.00000 0.00000 1.00000 Fe1a 0.01033 I XU MMM 4 000
3.500 0.000 0.000
4 FE+3 0.00000 0.00000 0.50000 1.00000 Fe1b 0.01033 I XU MMM 4 000
-3.500 0.000 0.000
5 FE+3 0.00000 0.00000 0.16943 1.00000 Fe2a 0.01033 I XU MM2(001) 8 000
-3.500 0.000 0.000
6 FE+3 0.00000 0.00000 0.33057 1.00000 Fe2b 0.01033 I XU MM2(001) 8 000
3.500 0.000 0.000
7 O 0.00000 0.00000 0.08755 1.00000 O1a 0.01789 I XU MM2(001) 8 000
8 O 0.00000 0.00000-0.41245 1.00000 O1b 0.01789 I XU MM2(001) 8 000
9 O 0.25000 0.00000 0.19336 1.00000 O2 0.01789 I XU M(010) 16 000
10 O 0.00000 0.25000 0.18982 1.00000 O3 0.01789 I XU M(100) 16 000
11 O 0.00000 0.25000 0.00000 0.86368 O4 0.01789 IFXU MM2(010) 8 000
12 O 0.25000 0.00000 0.00000 1.06416 O5 0.01789 IFXU MM2(100) 8 000
Phase No. 1; Phase has 12 atoms; Title: from /Users/toby/proj/ybafe/YBaFeO_
Give atom editing command
(,$,C,D,E,F,I,K,L,M,P,S,T,U,V,X,+,-,*,/) >x x x x
EXPEDT data setup option (,D,F,K,L,P,R,S,X) >
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Check Magnetic Form Factor | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
For this example, GSAS already has the correct magnetic form factor loaded for atom type FE+3, but it is always a good idea to check this. In some cases, the magnetic form factor for a problem will need to be loaded. Many magnetic form factors can be found in the International Tables of Crystallography, Volume C in section 4.4.5. Note that GSAS uses the terms A1, B1, A2, B2, A3, B3, and C for the terms labeled A, a, B, b, C, c and D in the International Tables. GSAS terms A4 and B4 are not used in the International Tables and should be specified as zero. In EXPEDT, use the following commands:
Note that if EXPGUI were to be started at this point, the first command would be "Y" to indicate that the correct file has been selected to be edited. Command 1) [L] enters the least-squares menu; command 2) [F] moves from the least-squares to the form factor submenu; command 3) [M FE+3] opens editing of the magnetic form factor for Fe3+ command 4) [C] specifies that the saved values will be changed, the values [.3972 13.244 .6295 4.903 -.0313 .35 0 0 .0044] specify the new <j0> coefficients. command 5) [n] indicates that the <j2> terms will not be changed. command 6) [U] specifies that the entered coefficients are to be saved. If this U is not entered, the original values will not be changed and the new values are ignored -- use care to remember to enter this command. Command 7) [x x x x] returns to the main menu. The input to and output from EXPEDT are shown below. User-typed input is emphasized by display in this font: Input
EXPEDT data setup option (,D,F,K,L,P,R,S,X) >L Select editing option for Least Squares calculation (,A,B,F,H,L,O,R,S,T,X) >F Form factor editing options - (,M,N,R,S,X) >M Enter atom type >FE+3 F-factor is expressed in the form : A(I)*EXP(-B(I)*SQ) + C , I=1 to 4 F-factor is expressed in the form : [A(I)*EXP(-B(I)*SQ) + C]*SQ , I=1 to 4 where SQ is (Sin theta/lambda)**2 Magnetic form-factor coeffs. for FE+3 are A1,B1,A2,B2 = 0.39720 13.24400 0.62950 4.90300 A3,B3,A4,B4,C= -0.03140 0.35000 0.00000 0.00000 0.00440 mff(0) = 0.99970 Magnetic form-factor coeffs. for FE+3 are A1,B1,A2,B2 = 1.36020 11.99800 1.51880 5.00300 A3,B3,A4,B4,C= 0.47050 1.99100 0.00000 0.00000 0.00380 Lande g factor = 2.00 Magnetic form factor editing options - (,A,C,E,G,L,P,R,U,X) >C Enter A(1) & B(1) >.3972 13.244 Enter A(2) & B(2) >.6295 4.903 Enter A(3) & B(3) >-.0313 .35 Enter A(4) & B(4) >0 0 Enter C >.0044 Do you want to modify the values (Y/)? >n F-factor is expressed in the form : A(I)*EXP(-B(I)*SQ) + C , I=1 to 4 F-factor is expressed in the form : [A(I)*EXP(-B(I)*SQ) + C]*SQ , I=1 to 4 where SQ is (Sin theta/lambda)**2 Magnetic form-factor coeffs. for FE+3 are A1,B1,A2,B2 = 0.39720 13.24400 0.62950 4.90300 A3,B3,A4,B4,C= -0.03130 0.35000 0.00000 0.00000 0.00440 mff(0) = 0.99980 Magnetic form-factor coeffs. for FE+3 are A1,B1,A2,B2 = 1.36020 11.99800 1.51880 5.00300 A3,B3,A4,B4,C= 0.47050 1.99100 0.00000 0.00000 0.00380 Lande g factor = 2.00 Magnetic form factor editing options - (,A,C,E,G,L,P,R,U,X) >U Magnetic form factor editing options - (,A,C,E,G,L,P,R,U,X) >x x x x EXPEDT data setup option (,D,F,K,L,P,R,S,X) >x STOP EXPEDT terminated successfully statement executed Press Enter to continue
After the EXPEDT window is closed since EXPEDT has changed the experiment file, (unless the Autoload EXP option is set) there is a prompt to load the information from this changed file: press the "Load new" button to continue. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine with Magnetic Phase | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
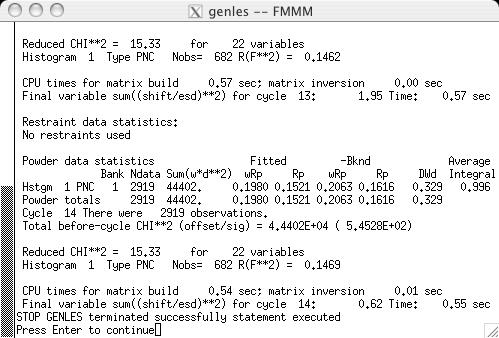
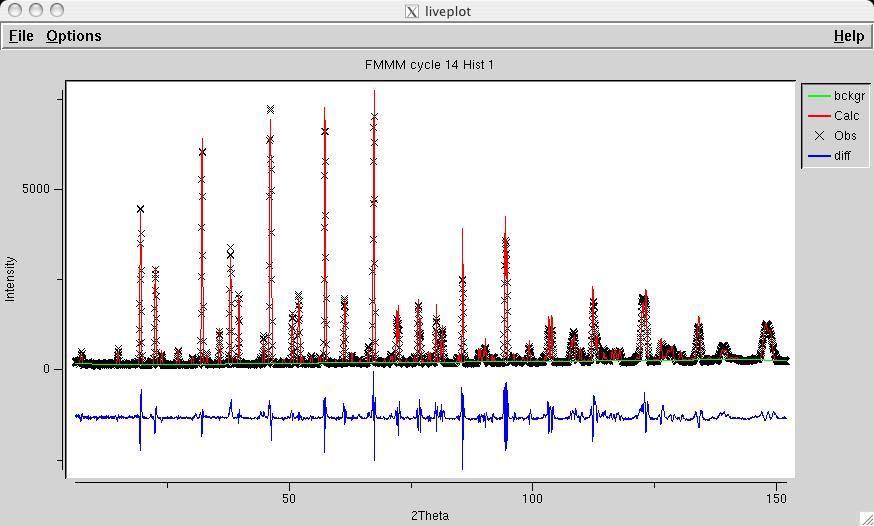
Since the phase type has been changed, it is necessary to run POWPREF (screen image). and then GENLES (screen image). The fit improves from the previous chi2 value of approximately 21 to a new value of approximately 15, as the low-angle magnetic lines are now computed with more intensity, as can now be seen in LIVEPLOT (screen image). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine Profile Constraints | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
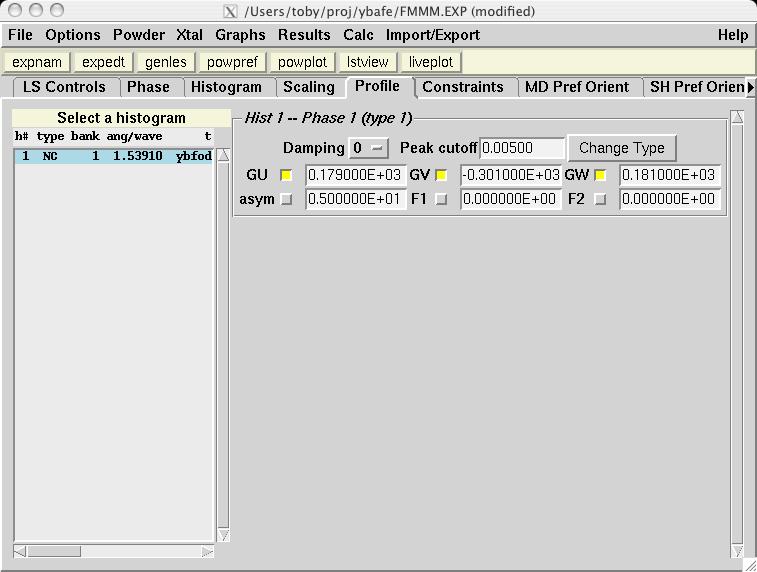
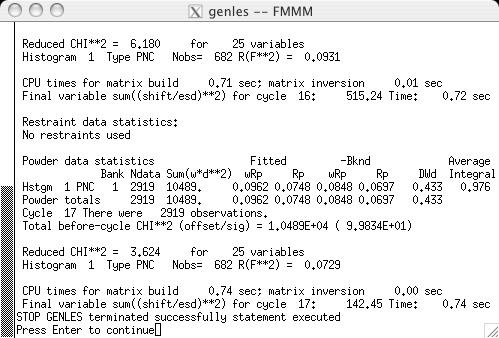
From the previous LIVEPLOT result, it is clear that the profile is a major source of differences between the observed and computed diffraction data. While it is possible to have different ordering ranges for the nuclear and magnetic ordering, this cannot be incorporated into a refinement with a single nuclear and magnetic phase. Select the check buttons for GU, GV & GW on the Profile panel (screen image) and then start GENLES (screen image). The fit improves from the previous chi2 value of approximately 15 to a new value of approximately 3.4. Running POWPREF and then GENLES again causes chi2 to drop from 3.4 to 2.8 just due to the expansion of the peak shape range from POWPREF. Subsequent refinement cycles improves chi2 slightly, to 2.6. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Constrain & Refine Magnetic Moment | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
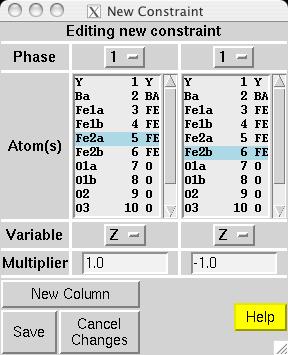
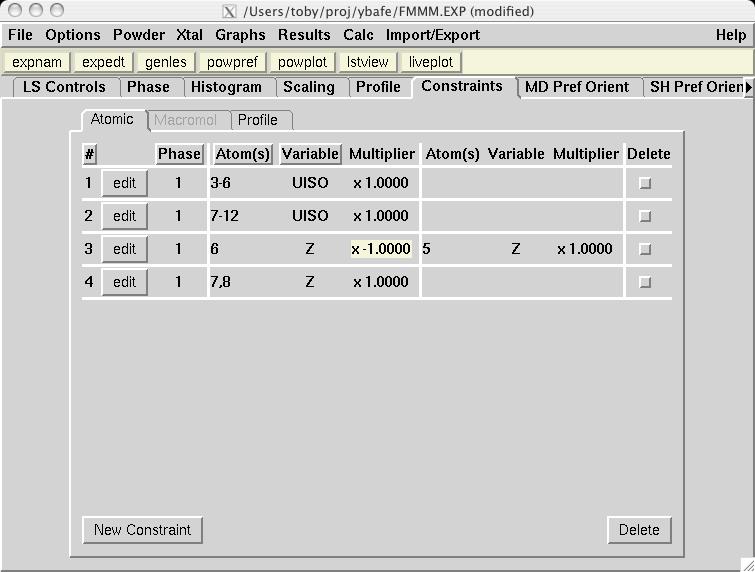
|
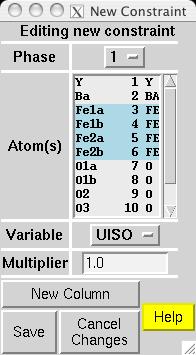
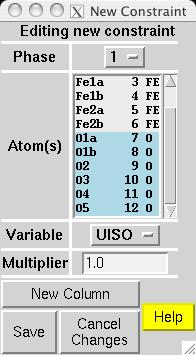
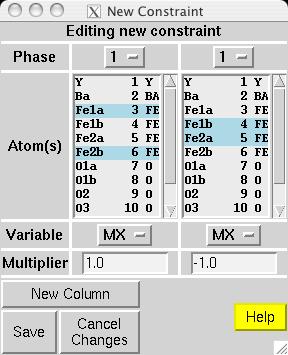
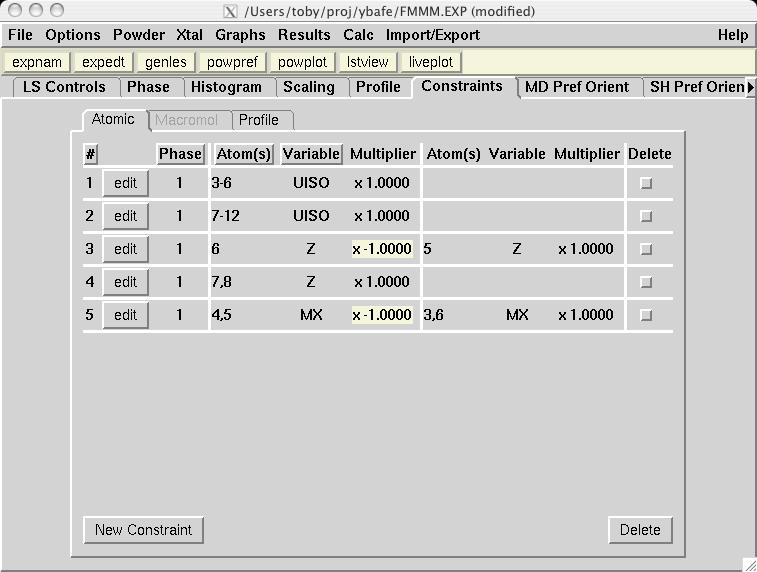
In most magnetic refinements, one wishes to refine magnetic moments. It should be noted that the Fe1a and Fe1b atoms are symmetry related, as is the case for Fe2a and Fe2b. One could consider treating the two Fe1 atoms as having different moments from the two Fe2 atoms, but this makes little chemical sense, since there is no reason to expect either site to have an accumulation of off-valence Fe atoms. Further, due to the pseudo-symmmetry of the cell, there are few reflections that distinguish the two non-equivalent Fe scatterers. Thus, it is best to treat all Fe atoms as having a single magnetic moment. EXPGUI can be used to set up constraints on the magnetic moments, by creating a new atom constraint on the Constraints panel, where a shift multiplier of 1.0 is applied to MX of atoms 3 & 6 and a shift multiplier of -1.0 is applied to MX of atoms 4 & 5 (screen image). Press the "Save" button and the constraint appears (screen image). Changing the refinement flag for the magnetic moments must currently be done from EXPEDT, using the following commands:
where command 1) [Y] indicates the correct file has been selected to be edited; [L] enters the least-squares menu; command 2) [A] moves from the least-squares to the atoms submenu; command 3) [v 3:6 M] sets the M flag (refine magnetic moment) for atoms 3 through 6 (the 4 Fe atoms) command 4) [x x x x] returns to the main menu; command 5) [x] exits EXPEDT The input to and output from EXPEDT are shown below. User-typed input is emphasized by display in this font: Input
|------------------------------------------|
| Program EXPEDT Version MacOSX |
| A menu driven routine to edit .EXP files |
| Distributed on Thu Sep 30 14:58:36 2004 |
|------------------------------------------|
|---------------------------------------------------------------|
| Allen C. Larson and Robert B. Von Dreele |
| Manuel Lujan, Jr. Neutron Scattering Center, MS-H805 |
| Los Alamos National Laboratory, Los Alamos, NM 87545 |
| |
| Copyright, 2000, The Regents of the University of California. |
|---------------------------------------------------------------|
The last history record is :
HSTRY 24 EXPGUI 1.74 1.42 (1 changes) -- 07/30/05 22:57:33
Is this the file you wish to use? (,D,K,Q,R,Y) >Y
Experiment title:
Model nuclear & magnetic scattering in Fmmm
The last history record is :
HSTRY 24 EXPGUI 1.74 1.42 (1 changes) -- 07/30/05 22:57:33
EXPEDT data setup option (,D,F,K,L,P,R,S,X) >l a
Phase No. 1; Phase has 12 atoms; Title: from /Users/toby/proj/ybafe/YBaFeO_
Give atom editing command
(,$,C,D,E,F,I,K,L,M,P,S,T,U,V,X,+,-,*,/) >v 3:6 M
SER TYPE X Y Z FRAC NAME UISO CODE STSYM MULT FXU
MX MY MZ MCODE
3 FE 0.00000 0.00000 0.00000 1.00000 Fe1a 0.00655 I XU MMM 4 000
3.500 0.000 0.000 M
4 FE 0.00000 0.00000 0.50000 1.00000 Fe1b 0.00655 I XU MMM 4 000
-3.500 0.000 0.000 M
5 FE 0.00000 0.00000 0.17002 1.00000 Fe2a 0.00655 I XU MM2(001) 8 000
-3.500 0.000 0.000 M
6 FE 0.00000 0.00000 0.32998 1.00000 Fe2b 0.00655 I XU MM2(001) 8 000
3.500 0.000 0.000 M
Phase No. 1; Phase has 12 atoms; Title: from /Users/toby/proj/ybafe/YBaFeO_
Give atom editing command
(,$,C,D,E,F,I,K,L,M,P,S,T,U,V,X,+,-,*,/) >x x x x
EXPEDT data setup option (,D,F,K,L,P,R,S,X) >x
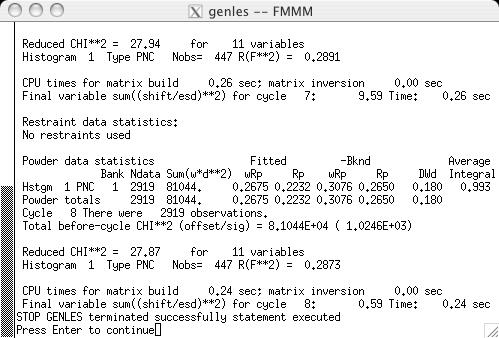
The refinement is then started with GENLES. The magnetic moments shift only slightly (as can be seen from the LSTVIEW output) and the chi2 value drops only slightly (screen image). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Further Model Improvements | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
At this point the refinement has progressed significantly, but for publication one might wish to obtain a slightly better fit. To do this, the number of background parameters can be increased to the point where little improvement is seen by adding more terms, say to around 12-14 terms. The profile can be improved further by switching to profile type 2 or 3 and allowing the LX term to refine. There is a minor impurity phase, BaFeO3, that can be included (Pm3m, a=4.0875, Ba: 0,0,0; Fe 1/2,1/2,1/2; O 1/2,1/2,0). Note that the phase fraction for this phase can be refined, and later the unit cell, but the Uiso should be fixed at a reasonable level, such as the default of 0.025. With these additions, the fit can be improved to a reasonable chi2 = 2.1 and very good R(F2) = 0.044. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}