
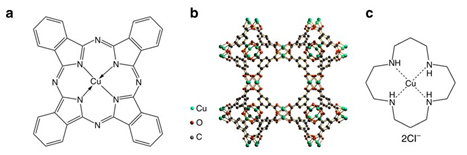
Molecular structures of three Cu-complex materials and their electrocatalytic performance for CO2 reduction. Molecular structures of a CuPc, b HKUST-1, and c [Cu(cyclam)]Cl2. Potential-dependent d Faradaic efficiencies and e partial current densities of products for CO2 electroreduction reaction catalyzed by CuPc. Comparison of f Faradaic efficiency and g partial current density distributions among CO2 electroreduction reactions catalyzed by the three materials at – 1.06 V vs RHE. Error bars represent the SD from multiple measurements.
A unique phenomenon — reversible restructuring of metal-complex structures that enables high electrocatalytic performance — has been discovered by researchers employing high-brightness x-rays to carry out structural characterization of samples at the U.S. Department of Energy’s Advanced Photon Source (APS) at Argonne National Laboratory. This work, published in Nature Communications, has the potential for positive impacts in the realms of energy conservation and industrial chemistry.
In theory, electrocatalysts can be used to convert the ever-growing carbon dioxide (CO2) in the Earth’s atmosphere to carbon-neutral fuels or other value-added chemicals. Doing so on a large scale would diminish the environmental problems associated with the atmospheric CO2 and also benefit energy storage and chemical production. One attractive proposal is to power catalytic CO2 conversion with electricity generated from renewable energy sources, such as wind or solar energy. This electrocatalytic CO2 reduction would work under ambient conditions in aqueous media. However, this process requires cost-effective electrocatalysts for the reaction. The research team in this study led by Yale University, Oregon State University, and South University of Science and Technology had earlier discovered a promising candidate, a copper porphyrin, which is able to convert CO2 to methane in a neutral aqueous electrolyte.
It is known that the structures of many catalysts change under reaction conditions. This restructuring can be caused by various conditions, including temperature, pressure, electrical potential, and chemical environment. The structures formed during the reaction conditions are responsible for the resulting catalytic properties.
Using in situ and operando x-ray absorption spectroscopy (XAS) at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) beamline 5-BM-D at the APS, the team was able to follow the restructuring that occurred under CO2 reduction conditions in an aqueous electrolyte for three copper catalysts: copper(II) phthalocyanine (CuPc), copper(II) benzene-1,3,5-tricarboxylate (HKUST-1), and copper(II) 1,4,8,11-tetraazacyclotetradecane ([Cu(cyclam)]Cl2) (Fig. 1). (The APS is an Office of Science user facility at Argonne National Laboratory.)
Samples were prepared for analysis by drop drying an ink solution containing the catalysts on carbon fiber paper (2.5 by 1.5 cm2). Supplementary analyses of the samples included scanning electron microscopy, x-ray diffraction, and electrocatalytic measurements.
The authors correlated the catalyst structures determined from the XAS and other data to the observed catalytic properties. Of the three electrocatalysts, CuPc was clearly the most efficient for electrochemical reduction of CO2 to methane (66% Faradaic efficiency) and attained the highest current density (13 mA/cm2). The XAS structural data showed that during the conversion reaction the CuPc molecules restructure to metallic copper clusters (size of around 2 nm), which are the active sites for the electrocatalysis (Fig. 2). Then, the copper nanoclusters reverse back to the original CuPc structure after the negative electrode potential is removed. By contrast, the other two samples decompose to much larger copper nanostructures, and do not return to the original structure. The excellent performance of the CuPc electrocatalyst is due to its reversible formation of the copper nanoclusters. The earlier reported copper porphyrin catalyst shows a similar restructuring behavior as CuPc.
The authors also performed density functional theory calculations on the restructuring of the CuPc material. These calculations revealed that the causes of its good reversibility were the small size of the copper nanoclusters and the strong affinity of the copper ions and phthalocyanine ligands.
Co-author Hailiang Wang (Yale University) commented that “The results from our study provide valuable insight into strategies for developing other high-performance electrocatalysts.” His team is developing other heterogenized molecular catalysts with new functionalities and high performance for electrochemical CO2 conversion. — Joseph E. Harmon
See: Zhe Weng1,2, Yueshen Wu2, Maoyu Wang3, Jianbing Jiang2, Ke Yang2, Shengjuan Huo2,4, Xiao-Feng Wang5, Qing Ma6, Gary W. Brudvig2, Victor S. Batista2, Yongye Liang1*, Zhenxing Feng3**, and Hailiang Wang2***, “Active sites of copper-complex catalytic materials for electrochemical carbon dioxide reduction,” Nat. Commun. 9, 415 (2018). DOI: 10.1038/s41467-018-02819-7
Author affiliations: 1South University of Science and Technology of China, 2Yale University, 3Oregon State University, 4Shanghai University, 5University of South China, 6Northwestern University
Correspondence: *[email protected], **[email protected], ***[email protected]
The work was supported by the National Science Foundation (Grant CHE-1651717), a Doctoral New Investigator grant from the ACS Petroleum Research Fund, a Global Innovation Initiative from the Institute of International Education, and the Callahan Faculty Scholar Endowment Fund from Oregon State University. The synthetic work was supported by the U.S. Department of Energy (DOE), Chemical Sciences, Geosciences, and Biosciences Division, Office of Sciences–Basic Energy Sciences (Grant DEFG02-07ER15909). Additional support was provided by a generous donation from the TomKat Foundation. V.S.B. acknowledges support from the Air Force Office of Scientific Research grant FA9550-13-1-0020 and supercomputing time from the NERSC. Y.L. acknowledges financial support from Shenzhen Fundamental Research Funding (JCYJ20160608140827794) and Peacock Plan (KQTD20140630160825828). DND-CAT is supported by Northwestern University, E.I. DuPont de Nemours & Co., and The Dow Chemical Company. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.