
Hydrogen gas (H2) is used in a variety of commercial applications, including fertilizer production and as fuel for automotive and industrial fuel cells. Most H2 is extracted from hydrocarbons, especially via steam reforming of natural gas and the gasification of coal. These extraction processes produce a hydrogen/carbon monoxide (H2/CO) mix referred to as “synthesis gas.” A long-established catalytic reaction called the “water-gas shift” removes CO from synthesis gas by combining CO with water to form additional hydrogen plus CO2.
Now, a research team has unveiled a promising catalyst for low-temperature and low-cost water-gas shift processing. X-ray measurements performed at the U.S. Departmrnt of Energy's Advanced Photon Source (APS), an Office of Science user facility,were used to probe the distribution of various forms of the catalyst under real-world operating conditions. The new catalyst holds considerable potential for producing less-expensive alternative fuels, and for removing CO impurities from hydrogen for fuel cells. But perhaps the most far-reaching implication of this research is that current catalytic systems employing nanoparticles of precious metals may be overusing these expensive and rare materials, since catalysis is actually taking place at the scale of single atoms. This finding could lead to more efficient utilization of precious metals in the catalysts of the future. Catalysts with single-precious-metal-centers may also find use in other fuel processing reactions, and in the production of chemicals.
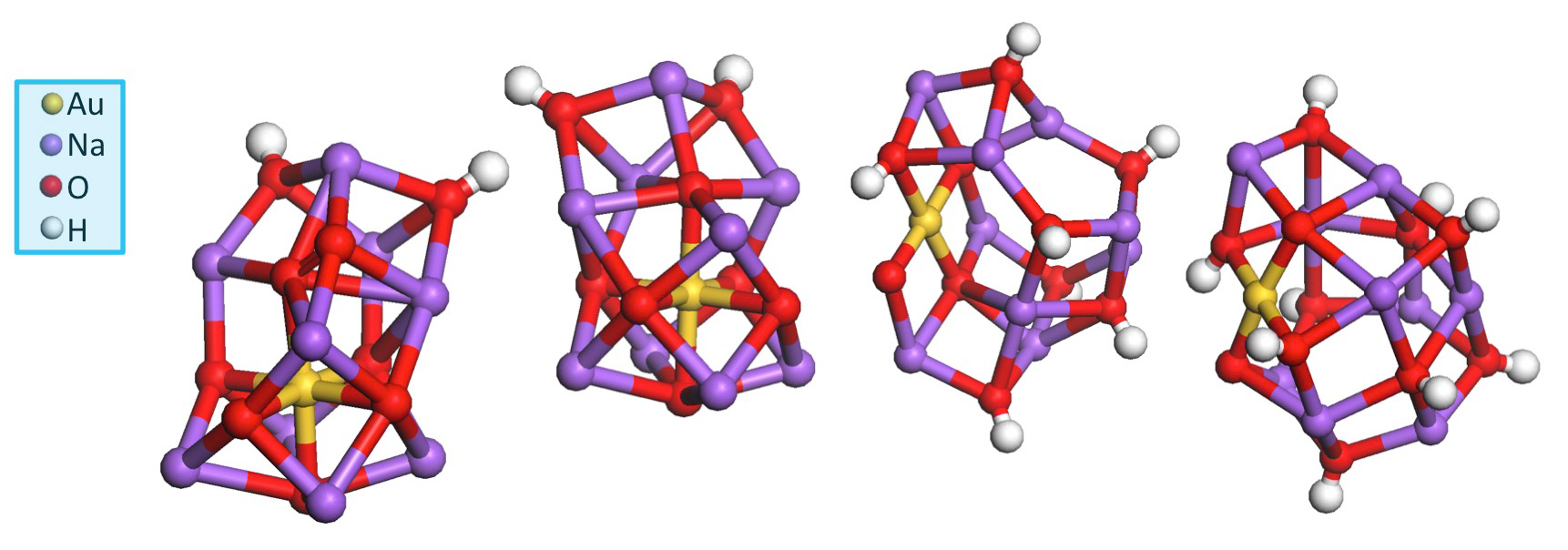
The researchers from Tufts University, the University of Wisconsin–Madison, Louisiana State University, Oak Ridge National Laboratory, Argonne, and the University of Sydney (Australia) found that each catalytic site is composed of a single gold atom attached to peripheral sodium or potassium ions through oxygen bonds. The gold-centric clusters can be adsorbed on a variety of supports, including zeolites and mesoporous silica, and are stable over a range of catalytic temperatures.
Although many catalysts can perform the water-gas shift reaction, each has certain limitations. Copper oxide, for instance, is widely used by industry to catalyze the water-gas shift. However, copper is pyrophoric, meaning it can spontaneously ignite in air at relatively low temperatures unless properly protected. This limits copper's usefulness with automotive fuel cells. Although there are non-pyrophoric catalysts, they typically have other drawbacks, such as requiring higher operating temperatures than gold, or using excessive amounts of expensive and/or scarce carrier materials (i.e., the materials supporting the catalyst).
The new gold-based catalyst avoids these problems. It is non-pyrophoric, and it provides a sustainable solution to CO removal: by incorporating just a single gold atom within each catalytic site, utilization of the precious metal is maximized. Moreover, the alkali metals used in the gold-centric catalyst are abundant and inexpensive, as are the silica-based and other non-exotic carriers. The first figure illustrates four computationally-derived catalytic clusters that best fit the actual structures of the catalytic sites produced in this research.
This single-gold-atom catalyst is based on a similar platinum catalyst developed previously by the same Tufts University team that led this research. The original platinum-based catalyst features a single platinum atom combined with oxygen, hydroxide, and sodium/potassium. The researchers substituted gold for platinum since gold has a lower activation energy, Ea, which yields a lower catalytic temperature (<200º C [~400º F]). To the surprise of the researchers, the substitution succeeded, with gold performing well in place of platinum.
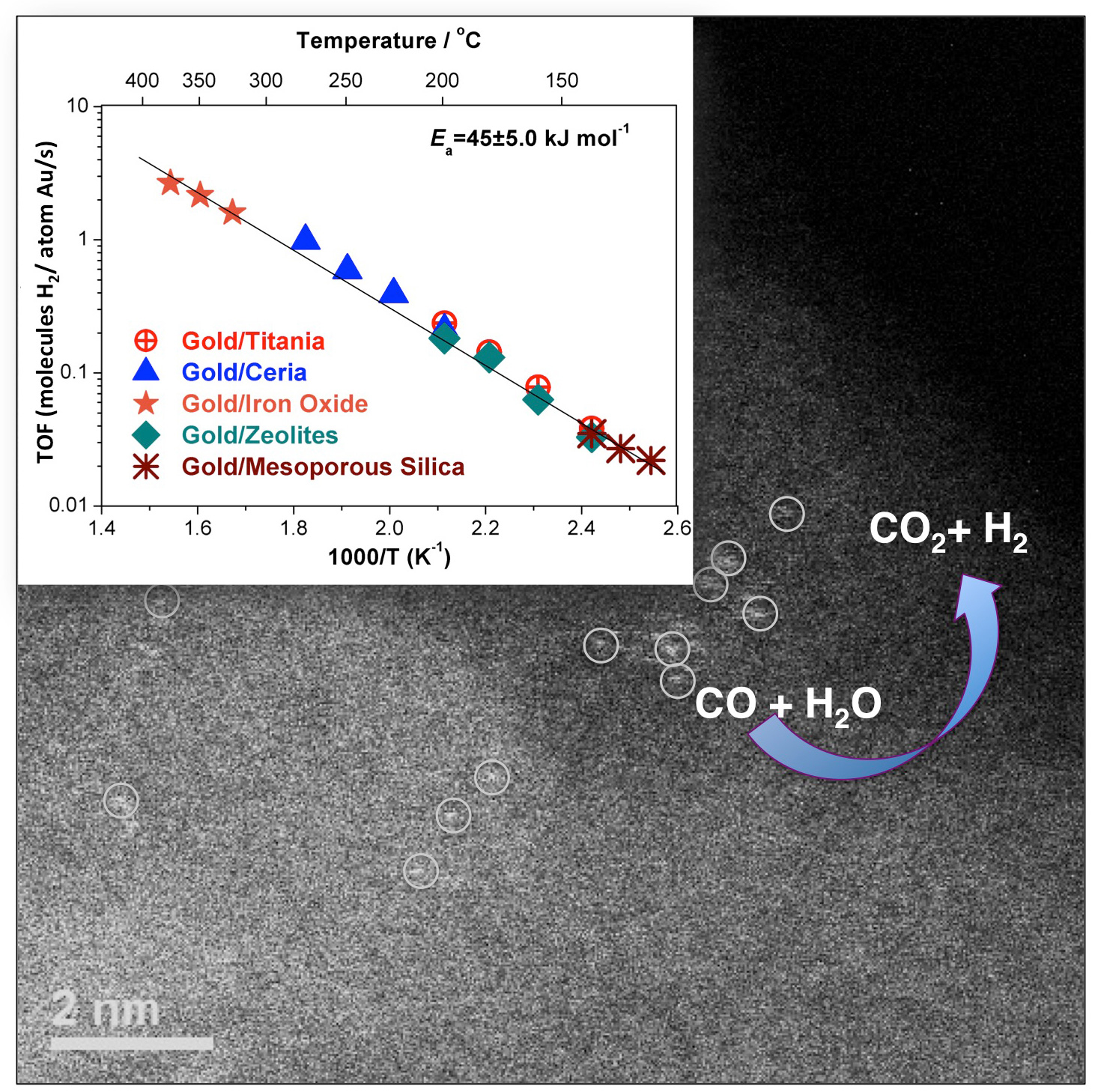
To gauge the effects of adding alkali ions to the atomic-gold catalyst, different samples were prepared, with some containing potassium or sodium ions, and some not. Each sample was subsequently dispersed into a silica-based carrier. Electron microscopy and various x-ray techniques were then used to probe the distribution of the catalysts within the carrier. Electron micrographs (see the second figure) indicate that the atomic-gold catalysts containing alkali ions were thoroughly distributed throughout their carriers, with no clumping observed.
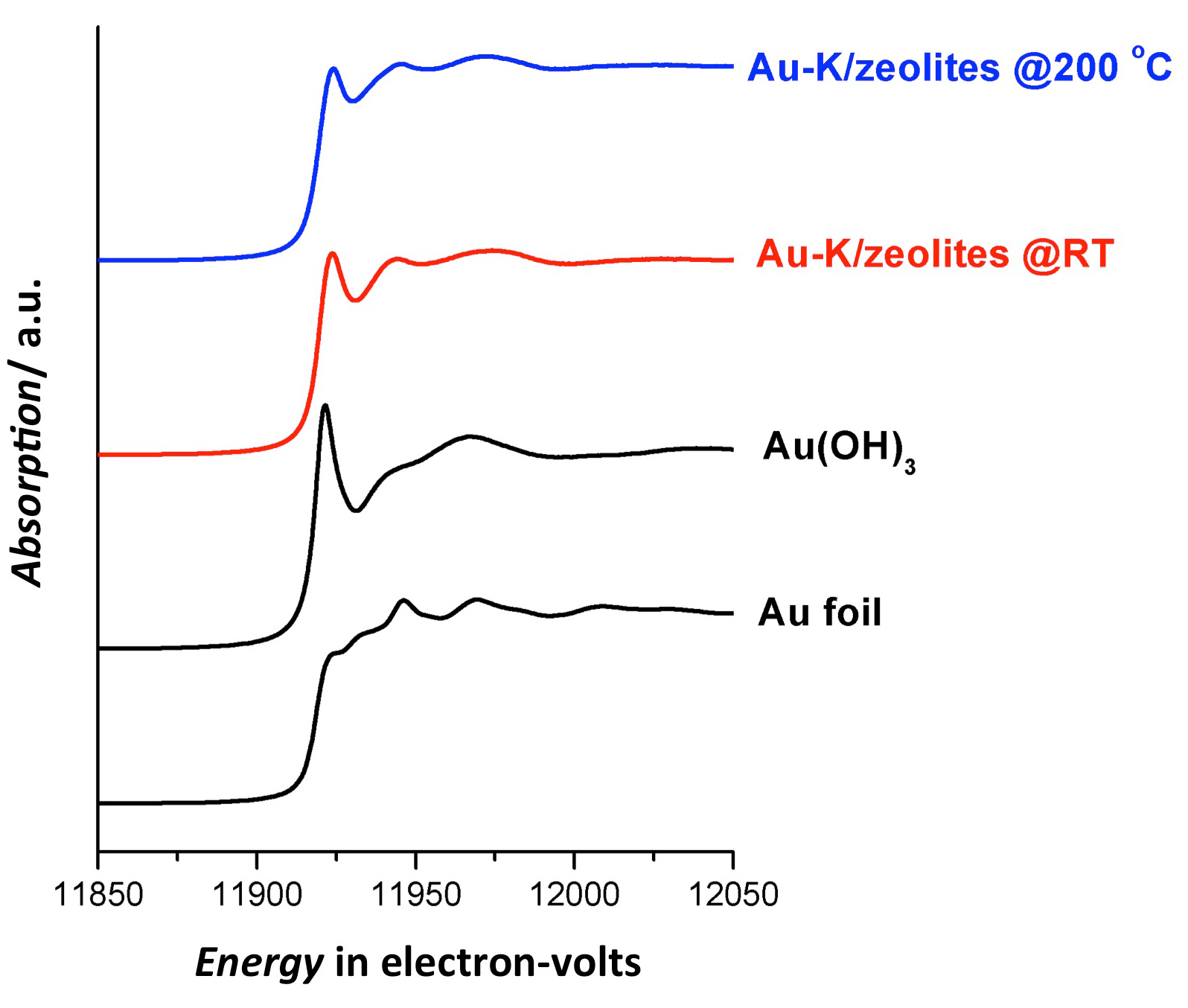
X-ray absorption spectroscopy (XAS) was likewise used to probe the distributions of the atomic-gold catalysts within the silica-based carriers. The x-ray measurements were performed at the X-ray Science Division x-ray beamline 12-BM-B of the APS at Argonne National Laboratory. The XAS data on the atomic-gold catalysts were collected while their carrier materials were subjected to conditions mimicking an actual water-gas shift conversion, including prolonged exposure to a CO/H2O mixture at catalytic temperatures (see the third figure). The XAS results confirmed the excellent distribution of the alkali-stabilized gold catalysts within the carriers, with no observed metallic aggregation, including the complete absence of nanoparticle formation. Conversely, both electron microscopy and XAS revealed that the atomic-gold catalysts lacking alkali ions did not disperse well within the supports, but instead formed nanoparticles that suppressed their catalytic activity.
The new alkali-stabilized atomic gold catalysts, dispersed within silica carriers, demonstrated stability in excess of four days (100 h) under catalytic conditions. Moreover, they performed as well as conventional gold catalysts deposited on more-costly and less-abundant metal-oxide carriers.
— Philip Koth
See: Ming Yang1, Sha Li2, Yuan Wang1, Jeffrey A. Herron2, Ye Xu3, Lawrence F. Allard4, Sungsik Lee5, Jun Huang6, Manos Mavrikakis2, and Maria Flytzani-Stephanopoulos1*, “Catalytically active Au-O(OH)x species stabilized by alkali ions on zeolites and mesoporous oxides,” Science 346(6216), 1498 (19 December 2014).
DOI: 0.1126/science.1260526
Author affiliations: 1Tufts University, 2University of Wisconsin–Madison, 3Louisiana State University, 4Oak Ridge National Laboratory, 5Argonne National Laboratory, 6University of Sydney
Correspondence: *[email protected]
The financial support by the U.S. Department of Energy-Basic Energy Sciences (DOE-BES) under grant DE-FG02-05ER15730 is gratefully acknowledged. This research used resources of the Advanced Photon Source, a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Argonne National Laboratory is supported by the Office of Science of the U.S. Department of Energy. The Office of Science is the single largest supporter of basic research in the physical sciences in the United States, and is working to address some of the most pressing challenges of our time. For more information, please visit science.energy.gov.