
Antibiotics have saved millions of lives but have also been the cause of their own undoing. From the moment penicillin first started being used as an antibiotic in 1942, bacteria have been busily developing resistance to this life-saving drug and the others that have followed it. As we develop and use new antibiotics, bacteria develop resistance to them and we are forced into an arms race in which we must constantly develop new ways to battle these clever microscopic warriors. Penicillin was developed from a natural product that inhibits cell wall synthesis in bacteria. Although bacteria have found ways to circumvent the specific action of penicillin, the existence of other natural antibiotics that work by different but related mechanisms provides evidence that cell wall synthesis is still a good target for antibiotics. Now, results from the U.S. Department of Energy’s Advanced Photon Source (APS) have helped a team of researchers take an important step toward development of a new antibiotic that can block bacterial cell wall synthesis.
The focus of the team’s effort is the MraY enzyme, an integral membrane protein that catalyzes the first step in peptidoglycan biosynthesis. Peptidoglycan is an essential component of the bacterial cell wall and is important for cell division – the process that bacteria use to multiply. The researchers, from the Duke University Medical Center, Hokkaido University (Japan), and Duke University, solved the crystal structure of MraY in complex with a natural inhibitor, muraymycinD2 (MD2), that has shown the ability — in animal models and in the lab — to kill some of the most lethal antibiotic resistant bacteria, including methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus (VRE), and Mycobacterium tuberculosis. Understanding how MD2 inhibits the MraY enzyme is crucial for developing a similar compound for clinical use in people.
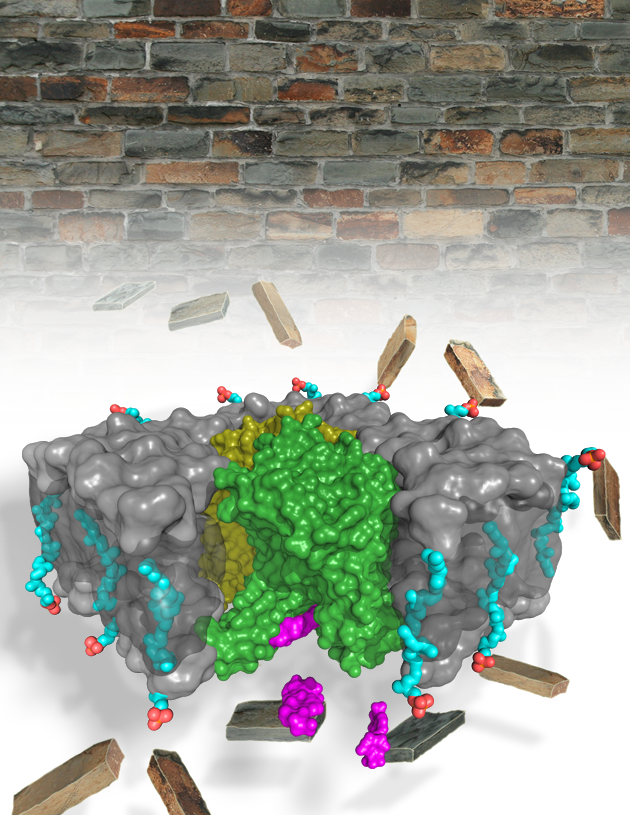
The structure, from x-ray data collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID-D and Northeastern Collaborative Access Team (NE-CAT) 24-ID-C beamlines at the APS, shows that MraY has 10 transmembrane helices (TMs) with 5 cytoplasmic loops. Most of the protein is in the membrane (Fig. 1). The catalytic site is a cleft framed by TM domains 3, 4, 5, 8, and 9, and loops B, C, D, and E. Three critical amino acids that are known to be important for catalysis and are conserved in this family of enzymes are located in the catalytic cleft. The APS is an Office of Science user facility.
One of the interesting questions about the MD2 inhibitor is how it can bind MraY even though it does not look anything like the natural substrate of the enzyme. Specifically, MD2 is a competitive inhibitor of MraY but does not have the sugar and pyrophosphate groups that are required for the natural substrate of MraY. The structure shows that MD2 inserts between loops C and D and interacts with TM9, but does not interact with the three critical catalytic amino acids. Instead, it causes a large conformational change in MraY in which TM9 rotates away from the active site and loop E rearranges. These changes reshape the active site and are accompanied by further rearrangements in loops A and D and in TM domains 1 and 5, and the unwinding of loop C. MD2 interacts with the new active site created by the movements of TM9 and loop E and with the new rearrangements of TM5 and loops C and D.
All of this moving around also creates substantial changes in the nature of the charges that are present in the active site, a potential factor in binding of the inhibitor. Interestingly, mutational analysis shows that the MD2 inhibitor does not interact with the same area of the active site as the part of the substrate it was previously thought to mimic, the pyrophosphate group, and in vitro experiments show that magnesium (Mg2+), which is critical for the enzymatic activity, is not important for MD2 binding.
The structure of MraY with its natural inhibitor, MD2, explains why MD2 does not need the same groups as the natural substrate in order to bind to the MraY active site and provides crucial information needed for designing drugs to inhibit this essential bacterial enzyme. — Sandy Field
See: Ben C. Chung1, Ellene H. Mashalidis1, Tetsuya Tanino2, Mijung Kim3, Akira Matsuda2, Jiyong Hong3, Satoshi Ichikawa2, and Seok-Yong Lee1*, “Structural insights into inhibition of lipid I production in bacterial cell wall synthesis,” Nature 533, 557 (26 May 2016). DOI: 10.1038/nature17636
Author affiliations: 1Duke University Medical Center, 2Hokkaido University, 3Duke University
Correspondence: *[email protected]
This work was supported by NIH R01 GM100984 (S.-Y.L.) and Duke startup funds (S.-Y.L.). This work was also supported by the JSPS Grant-in-Aid for Scientific Research on Innovative Areas “Chemical Biology of Natural Products” (S.I., grant number 24102502) and Scientific Research (B) (S.I., grant number 25293026). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Argonne National Laboratory seeks solutions to pressing national problems in science and technology. The nation's first national laboratory, Argonne conducts leading-edge basic and applied scientific research in virtually every scientific discipline. Argonne researchers work closely with researchers from hundreds of companies, universities, and federal, state and municipal agencies to help them solve their specific problems, advance America's scientific leadership and prepare the nation for a better future. With employees from more than 60 nations, Argonne is managed by UChicago Argonne, LLC for the U.S. Department of Energy's Office of Science.
The U.S. Department of Energy's Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, visit the Office of Science website.